
基于第一性原理的CL-20/C2H2/CH4燃烧的化学反应路径研究分享
文一:基于 FP-CL20 化学动力学模型对高能量密度材料 CL-20 的性能预测

高能密度材料(HEDM)的瞬时高能释放特征使它们成为高能推进剂或炸药的重要组成部分。因此,对HEDMs的性能的预测对其工程应用至关重要。在本文中,研究了Cl-20的爆炸反应过程,并分析了详细的化学反应动力学。结合量子化学计算,构建了第一个化学动力学模型(FP-CL20模型),其中包含153种和412个基本反应。通过使用FPCL20模型预测Cl-20爆炸性的热解和爆炸性能。在近似值的框架内,与实验结果预测的CL-20的关键物理量和爆炸的一致是令人满意的。FP-CL20模型还揭示了反应N2O+NO= NO2+N2和CO+NO2 =NO+CO2在爆炸下N2和CO2的形成中起关键作用。虽然与爆炸不同,但NCO+NO=N2+CO2和NCO+NO2=CO2+N2O是在热解下形成N2和CO2的主要反应。在爆炸反应区内,小分子N-heterochains(L-NCNCO+OH=NCN+HOCO)和小分子碳氧化物(HOCO+OH= CO2+H2O)的氧化是影响爆炸反应区时间的关键反应。我们的研究为了解Cl-20的热解和爆炸反应机理提供了一种新颖的见解,这也为化学动力学模型的构建和HEDM的性能预测铺平了道路。
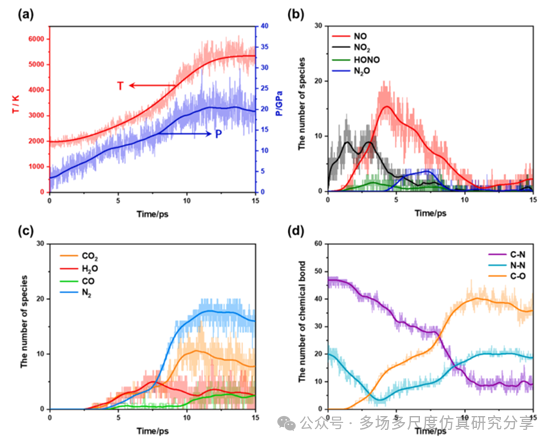
在2000 K时Cl-20热解中温度,压力,物种和化学键的时间演变。(a)温度和压力,(b)小分子中间体,(c)最终产物,(d)化学键。
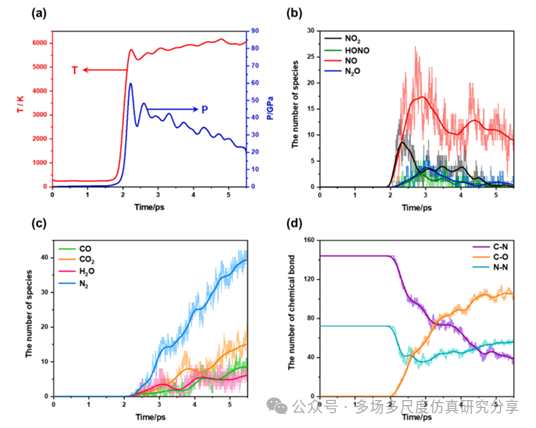
使用R-MSST在9 km/s的冲击下,温度,压力,物种和化学键在CL-20反应中的时间演变。(a)压力和温度,(b)小分子中间体,(c)最终产物,(d)化学键。
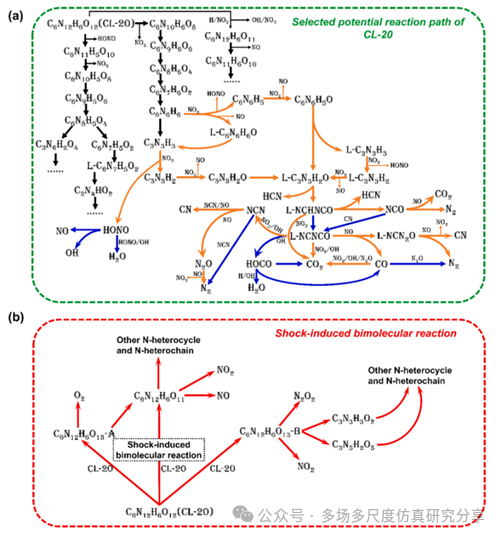
CL-20在热解和冲击下的选择性潜在反应路径。(a)热解和冲击下的联合化学反应路径。(b)冲击诱导的双分子反应。前缀“L-”代表作为N-杂链的结构,区别于N-杂环。
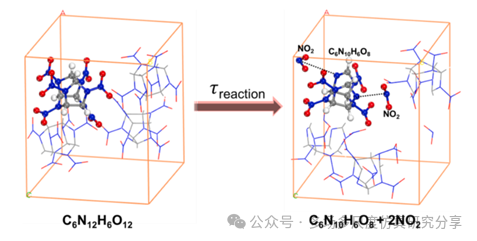
从CL-20的初始晶体结构到初始分解的反应快照。
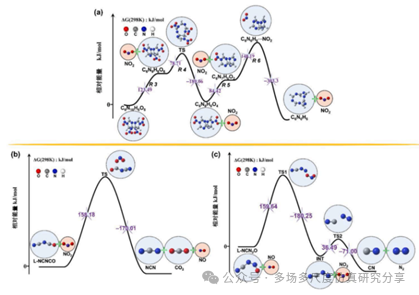
反应的势能面。(a) R 3, R 4, R 5 and R 6, (b) R 9, (c) R 10.
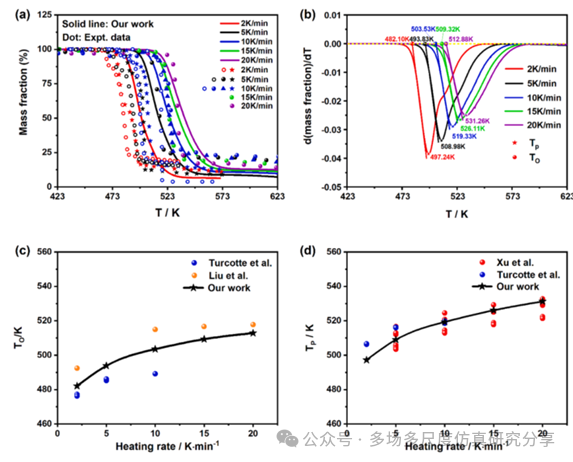
用FP-CL20模型预测不同升温速率下CL-20热解的TG曲线和DSC峰温。(a) TG曲线,(b) DTG曲线,(c)起始点火温度,(d) DSC峰值温度。为了表示开始点火温度,Tp表示峰值温度。DSC峰值温度的预测由预测的DTG峰值温度代替。
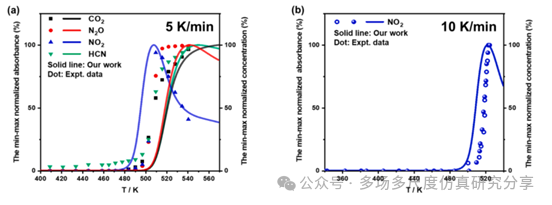
用FP-CL20模型预测不同升温速率下CL-20热解的组分浓度。(a) 5K/分钟,(b) 10K/分钟
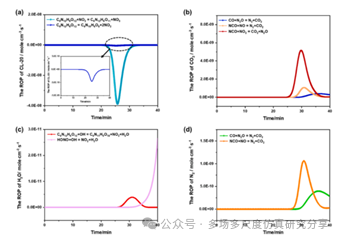
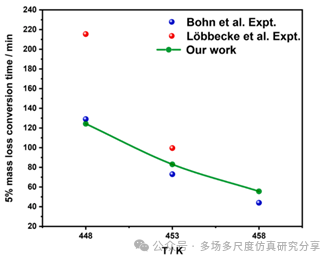
在5 K/min的加热速率下物种的ROP的时间演化。(a)氯-20,(b)二氧化碳,(c) H2O,(d) N2。用FP-CL20模型预测CL-20在448 K、453 K和458 K下热解的5 %质量损失转化时间。
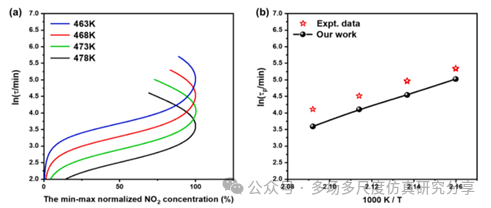
用FP-CL20模型预测CL-20在463 K、468 K、473 K和478 K下热解的NO2浓度峰值时间(τp)。(a) ln(τp)对NO2标准化浓度,(b) ln(τp)对1000/T。

CL-20炸药爆轰特征参数的预测。(a)物质浓度,(b)温度,(c)压力。

爆轰条件下物种ROP的时间演化。(a)氯-20,(b)二氧化碳,(c)H2O,(d)N2。
结论:基于第一性原理分子动力学方法,模拟了CL-20在热解和冲击下的反应。基于FPMD,我们发展了一种基于HEDMs的化学动力学模型构建方法。构建了第一个具有性能预测能力的CL-20化学动力学模型(FP-CL20模型)。通过数值模拟的热解和爆轰性能预测以及与实验的对比,验证了FP-CL20模型的准确性和适用性。具体结论如下:
1.CL-20的早期热分解主要是脱硝反应,而冲击下的早期CL-20反应主要是脱硝反应和冲击诱导的双分子反应。除反应前期外,反应中后期在CL-20的热分解和冲击下,反应路径相似。最终产物N2和CO2的形成主要与NO2和NO有关
2.结合量子化学计算,得到了153个物种和412个基元反应,并计算了基元反应的Arrhenius速率参数。首次建立了具有高精度性能预测能力的固体炸药CL-20化学动力学模型(FP-CL20模型)。
3.用FP-CL20模型模拟了实验条件下CL-20的热解。预测的TG曲线、DSC峰值温度、物种浓度、5 %质量损失转化时间、NO2浓度峰值时间等物理量与实验值一致。验证了FP-CL20模型描述CL-20热解动力学的准确性和适用性。根据ROP分析,低温热解中N2和CO2的生成主要与NCO和NO、NCO和NO2的双分子反应有关,如NCO+NO =N2+CO2和NCO+NO2 -- CO2+N2O。用FP-CL20模型预测炸药的热解性能也为评价炸药的热稳定性和贮存寿命提供了一种新的方法。
4.采用FP-CL20模型模拟了CL-20的爆轰反应。预测的爆压和爆轰反应区时间与实验值接近,验证了CL-20化学动力学模型在近似框架下描述CL-20爆轰反应动力学的准确性和适用性。根据ROP分析,爆轰条件下N2和CO2的快速形成主要与NO2的O迁移有关,如N2O+NO=NO2+N2、CO+NO2––NO+CO2等反应。此外,小分子氮杂链的氧化,如L-NCNCO+OH=NCN+HOCO,以及小分子碳氧化物的氧化,如HOCO+OH––CO2+H2O,是影响爆轰反应区时间和CJ压力、温度的关键反应。
总之,我们的研究为理解CL-20的热解和爆轰反应机理提供了新的视角,也为HEDMs化学动力学模型的构建和性能预测奠定了基础。
文二:对乙炔高温氧化动力学的深入研究:基于第一性原理的分子动力学模拟研究
乙炔(C2H2)高温氧化动力学及动力学模型的研究对其工程应用具有重要意义。本文首次用第一性原理分子动力学方法模拟了C2H2在高温下的氧化反应。结果表明,C2H2氧化过程中有38个中间体和225个基元反应。深入揭示了“瞬发”CO2以及气体污染物CHOCHO和HCOOH的形成机理。验证了目前动力学模型中争议较大的四种中间体CHCHO、CHOCO、CHOCHO和HCOOH。发现了一种新的中间产物CHOCO2。同时,我们的模拟也显示了自由基,如HO2,OH,O等。在C2H2氧化初期对中间体的氧化起关键的促进作用。结合量子化学计算,建立了C2H2/空气(FPC2H2)的详细化学动力学模型,并通过模拟着火延迟时间、流动反应器内组分浓度和预混层流火焰速度进行了验证。我们的研究为理解C2H2燃烧过程中空气污染物形成的复杂的化学反应动力学、环境和人类健康威胁。提供了新的视角。
C2H2/O2/Ar混合气点火延迟时间的六种数值模拟初始条件。
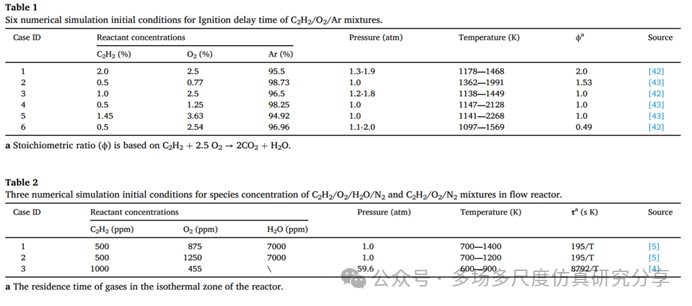
流动反应器中C2H2/O2/H2O/N2和C2H2/O2/N2混合物组分浓度的三种数值模拟初始条件。
(a)C2H2在O2中氧化的计算模型。(b)2000K时C2H2、H2O、CO2和CO总量的时间演变
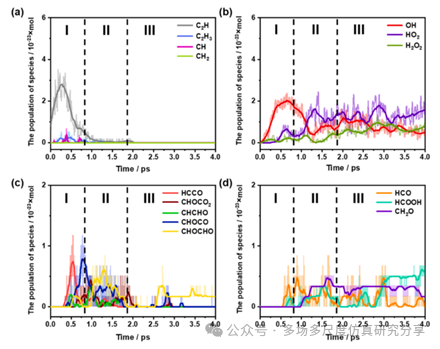
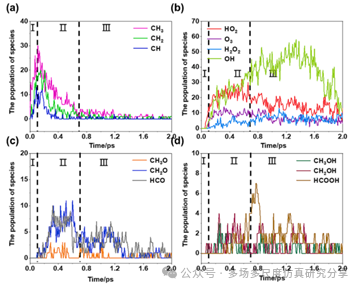
物种种群的时间进化。(a)含C-H的C1和C2物种。(b)含氢氧的物种。(c)含C2-氧化氢的物种。(d)含C1氢的物种。
C2H2生成CO2的主要反应途径和中间产物。CHCHO、CHOCHO、CHOCO、CHOCO2和HCOOH是新物种,它们被标记为蓝色。
关键基元反应的计算能垒和势能面。(a)与CHCHO和CHOCHO相关的基本反应。(b)与CHOCO和CHOCO2有关的基本反应。(c)与HCOOH相关的基本反应。(d)部分基元反应的势能面。
实验数据、模型预测了六种不同初始条件下预混C2H2/O2/Ar混合物的着火延迟时间。(a)案例1,(b)案例2,(c)案例3,(d)案例4,(e)案例5,(f)案例6。
FP-C2H2模型中CHOCO-,HCOOH相关反应在1400 K下着火时的温度敏感系数

FP-C2H2、USC II和Aramco 3.0模型中C2H2相关氧化反应在两种情况下着火时的温度敏感系数。
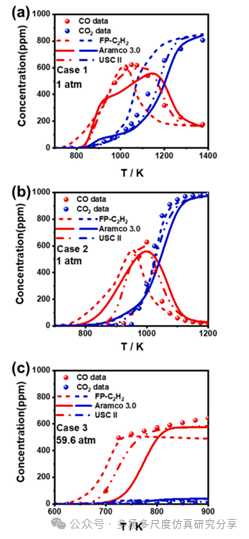
C2H2/ O2/H2O/N2和C2H2/O2/N2混合物在流动反应器中三种初始条件下氧化的模拟预测和实验数据的比较。浓度分布显示为反应器温度的函数。(a)案例1,(b)案例2。(c)案例3。点代表实验值,线代表来自不同模型的模拟值。
C2H2/空气混合物在两种初始条件下的层流火焰速度(a)案例1,(b)案例2。Tu代表未燃烧气体的温度。MRE值也显示在该图中。
利用FP-C2H2模型,在1 atm压力和1.2当量比下,研究了C2H2/空气预混层流火焰中HCCO、HCO、CO和CO2的ROP随温度的演化。(a) HCCO,(b) HCO,(c)一氧化碳,(d)二氧化碳。
结论:C2H2的燃烧化学反应动力学和动力学模型一直吸引着研究者的兴趣。本文用FPMD方法模拟了C2H2的高温氧化,得到了详细的基元反应和中间产物。探索了对C2H2化学动力学的认识。揭示了一些新的机理。建立并验证了C2H2/空气混合物的详细化学动力学模型FP-C2H2模型。主要结论如下:
1.发现了38个中间体和225个基元反应。燃烧化学动力学机理被详细理解。一些有趣的实验现象可以得到合理的解释。C2H2氧化的第一阶段中的CO和CO2“迅速”形成是由于CHOCO2 =HCO+CO2和CHOCO2 =H+CO+CO2。污染物乙二醛CHOCHO的形成可以表示为两步链增长序列:C2H2̅+O2→CHCHO̅+ O2 →CHOCHO。污染物HCOOH是由HCO+OH = HCOOH反应生成的。
2.验证了目前模型中存在差异的四种中间体CHCHO、CHOCO、CHOCHO和HCOOH。还发现了一种新的中间体CHOCO2。层流火焰的ROP分析和C2H2燃烧点火的温度敏感性分析揭示了它们的影响。CHOCO和HCOOH在C2H2燃烧过程中有重要作用。
3.在C2H2氧化的早期,自由基HO2、OH和O等。发挥关键作用。它们作为氧化剂促进含C-H中间体的氧化。
4.建立并验证了包含43个组分和239个反应的C2H2/空气燃烧化学动力学模型(FP-C2H2)。
在不同实验条件下,FP-C2H2模型对流动反应器中着火延迟时间、层流火焰速度和组分浓度的预测值与实验值一致。
文三:基于高精度第一性原理分子动力学模拟的甲烷/空气混合物燃烧的详细简化化学动力学模型
CH4/空气燃烧反应动力学因其在工程中的广泛应用而受到广泛关注。本文首次采用第一性原理分子动力学方法模拟了CH4在O2中的高温氧化反应。揭示了两种新的中间体HCOOH和O3及其对CH4氧化的影响。具体来说,将HCOOH相关反应加入到当前模型GRI 3.0、NUIG 1.1和USC II中可以显著改善它们对CH4/空气混合物的预混层流火焰速度的预测。基元反应分析还发现,OH、HO2等自由基在CH4氧化中起着关键作用。这些自由基作为高活性氧化剂,促进最终产物的形成。结合基元反应和简化技术,构建了一个新的第一原理(FP)和30步简化FP (R-FP)化学动力学模型。利用这两个模型,成功地预测了预混反应混合物(CH4/O2/Ar、CH4/O2/H2O/N2和CH4/空气)燃烧的着火延迟时间、流动反应器内组分浓度和预混层流火焰速度,并与实验结果进行了比较。总的来说,FP和R-FP模型有利于工程应用。
(a) CH4在O2中氧化的计算模型。(b) 4000时CH4、H2O、CO2和CO总量的时间演变
物种种群的时间进化。(a) CHX型中间体。(b) HXOY型和O3中间体。(c) CHXO型中间体。(d) CHXOYH型中间体。
(a)34种主要反应类型和主要基元反应。(b)二氧化碳的形成途径。(c)H2O的形成路径。
(a )-( i)HCOOH相关基元反应的Arrhenius动力学参数。白色、红色和灰色的球分别代表H、O和C原子。
R-FP模型的基元反应。
实验结果(Seery和Bowman,1970;斯帕达契尼和科尔凯特,1994年;Tsuboi和Wagner,1975)和预混反应混合物(CH4、O2和Ar)在九种不同初始条件下的点火延迟时间的预测值。
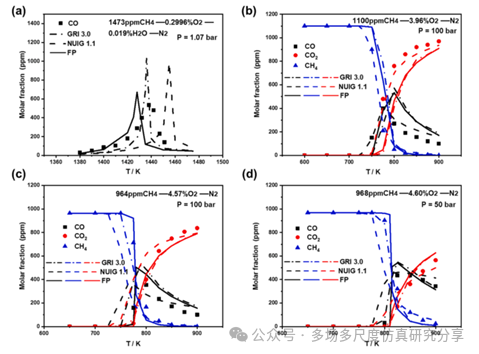
模拟预测和实验数据的比较(Smith等人,xxxx哈希米等人,2016;Rasmussen等人,2008年)在石英流反应器中的四种初始条件下的CH4/O2/H2O/N2混合物氧化。浓度分布显示为反应器温度的函数。(a)案例1,(b)案例2,(c)案例3,(d)案例4。
在1 atm和298 K的初始温度下,CH4/空气混合物的预混层流火焰速度。符号标记来自Iijima和Takeno (1986年)、Hassan等人(1998年)、Vagelopoulos和Egolfopoulos (1998年)的实验结果。在AAVR误差部分,G、U、N、G-S19分别代表GRI 3.0、USC II、NUIG 1.1和GRI S19模型。
CHXO型和CHXOYH型物种在1 atm和298 K的初始温度下的摩尔分数分布图(a) FP模型,当量比为0.8。(b) FP模型,当量比为1.2。(c) R-FP模型,当量比为0.8。(d) R-FP模型,当量比为1.2。
CH4/空气混合物在2、3、5、10 atm和298 K初始温度下的层流火焰速度。符号标记Hassan等人(1998年)、Rozenchan等人(2002年)、Egolfopoulos等人(1989年)、Gu等人(2000年)、Lowry等人(2011年)的实验结果。在part误差部分,G、U、N分别代表GRI 3.0、USC II、NUIG 1.1模型;G+、U+和N+分别代表m-GRI 3.0、m-USC II和m-NUIG 1.1型号。(a)情况1: 2个大气压,(b)情况2: 3个大气压,(c)情况3: 5个大气压,(d)情况4: 10个大气压。
用GRI 3.0模型和m-GRI 3.0模型在不同压力(1-20 ATM)和当量比(0.7-1.4)下预测CH4/空气预混层流火焰速度模拟中CO、HCO和HCOOH的摩尔分数峰值。(a)一氧化碳,(b) HCO,(c)羟基乙酸。
用m-GRI 3.0模型模拟CH4/空气预混层流火焰速度时不同压力和当量比下CO、CO2、HCOOH和HCO的ROP。(a)–( d)情况1:当量比为0.8,压力为1个大气压。(e)-(h)情况2:当量比为1.0,压力为1个大气压。(i)-(l)情况3:当量比为0.8,压力为5个大气压。(m)-(p)情况4:当量比为1.0,压力为5个大气压。
结论:本文用FPMD方法模拟了CH4在高温下的氧化过程。建立了CH4/空气混合物燃烧的详细化学动力学模型(FP模型)。结合DRG方法和CSP方法,得到了一个简化的反应化学动力学模型(R-FP ),该模型包括20种物种和30步反应。揭示了对CH4/空气燃烧机理的一些新认识。详细结论如下:
1.CH4在O2中氧化的模拟实验发现了22个中间体和123个基元反应。与目前的模型相比,发现了两种新的中间产物HCOOH和O3。反应机理表明,HCOOH主要影响CO和CO2的生成,O3影响OH自由基的生成。在目前已报道的模型(如GRI 3.0、USC II、NUIG 1.1)中加入HCOOH和HCOOH相关反应可以显著提高这些模型对CH4/空气预混层流火焰速度的预测,尤其是在低当量比(0.7-0.9)时。
2.自由基,如OH和O,在CH4氧化中起重要作用。这些自由基作为高活性氧化剂,促进最终产物的形成。此外,在简化的30步动力学模型中,80%的反应与OH、HO2、O自由基有关。
3.结合基元反应,建立了一个新的FP化学动力学模型。利用我们的FP模型,成功地预测了预混反应混合物(CH4/O2/Ar、CH4/O2/H2O/N2和CH4/空气混合物)燃烧的着火延迟时间、流动反应器中组分浓度和层流火焰速度,并与实验结果进行了比较。FP模型对层流火焰速度的预测值与实验数据吻合较好,尤其是在低当量比时。
4.基于FP模型建立了30步简化FP化学动力学模型,有利于复杂数值模拟在工程中的应用。
选自微信公众号 多场多尺度仿真研究分享